8 Merging CopyKit Objects
CopyKit objects can be merged into one object with the cbind() function.
Example:
# gain of chromosome 7 and deletion of chromosome 10
ck1 <- mock_bincounts(
ncells = 50,
ncells_diploid = 0,
position_gain = 4900:5493,
position_del = 6523:7056,
genome = "hg38",
resolution = "220kb"
)## Running variance stabilization transformation: ft## Smoothing outlier bins.## Running segmentation algorithm: CBS for genome hg38## Merging levels.## Done.# adding an identifier to colData
colData(ck1)$info <- 'object1'# gain of chromosome 7 and deletion of chromosome 10
# additional gain of chromosome 1
ck2 <- mock_bincounts(
ncells = 50,
ncells_diploid = 0,
position_gain = c(1:906, 4900:5493),
position_del = 6523:7056,
genome = "hg38",
resolution = "220kb"
)## Running variance stabilization transformation: ft## Smoothing outlier bins.## Running segmentation algorithm: CBS for genome hg38## Merging levels.## Done.# adding an identifier to colData
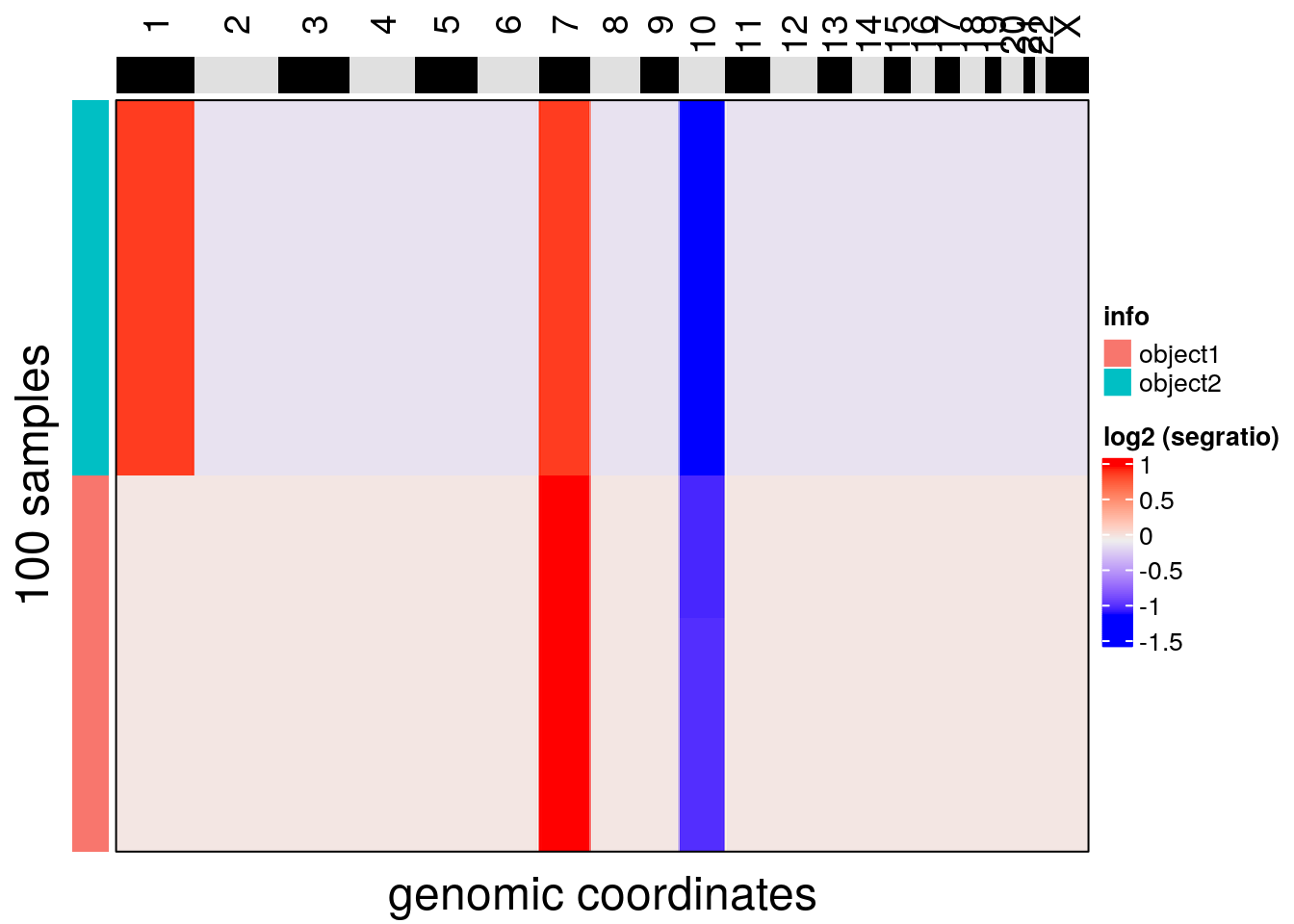
colData(ck2)$info <- 'object2'# merging objects
merged_copykit <- cbind(ck1, ck2)Following merge a standard CopyKit analysis can be applied
# UMAP and clustering
merged_copykit <- runUmap(merged_copykit)## Using assay: logr## Embedding data with UMAP. Using seed 17## Warning in .check_reddim_names(x, value, withDimnames): non-NULL 'rownames(value)' should be the same as
## 'colnames(x)' for 'reducedDim<-'. This will be an
## error in the next release of Bioconductor.## Access reduced dimensions slot with: reducedDim(scCNA, 'umap').## Done.merged_copykit <- findSuggestedK(merged_copykit)## Calculating jaccard similarity for k range: 10## ## Suggested k = 10 with median jaccard similarity of: 1merged_copykit <- findClusters(merged_copykit)## Using suggested k_subclones = 10## Finding clusters, using method: hdbscan## Found 2 subclones.## Done.plotUmap(merged_copykit, label = 'subclones')## Plotting Umap.## Coloring by: subclones.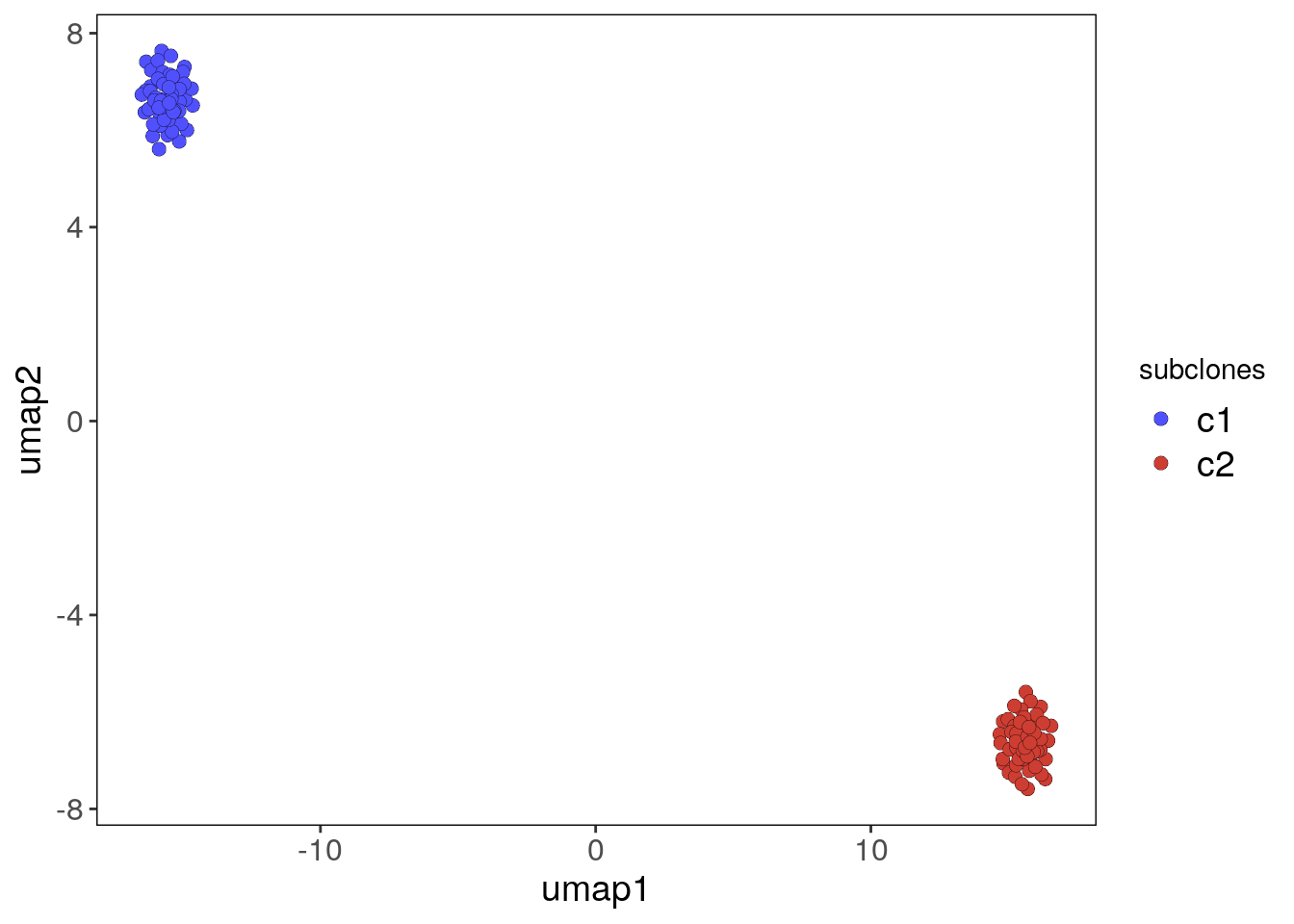
# plotting
plotHeatmap(merged_copykit, label = 'info', order_cells = 'hclust')## Ordering cells by:hclust## No distance matrix detected in the scCNA object.## Calculating distance matrix with metric: euclidean## Using 1 cores.## Access distance matrix with copykit::distMat()## Done.## Plotting Heatmap.